

A14 Assessing genetic and epigenetic heterogeneity in hematopoietic neoplasms using single cell genomics
While the evolutionary nature of cancer and hematopoietic neoplasms is well established, it is challenging to generate and analyze data to quantify the dynamics of this Darwinian process. This is a clinical problem, for example when therapy-resistant cancer cells relapse after an initially successful treatment. Hence, patients die because the tumor is able to evolve. To better understand and eventually influence this process, it is necessary to measure how much heritable variation exists and how fast it is generated, i.e. to measure the amount and rates of genetic and epigenetic heterogeneity. A decisive technological development to tackle this issue have been methods to characterize genetic and phenotypic heterogeneity at the single cell level. We have established such technologies (e.g. Fluidigm, SCRB-Seq, Drop-Seq), will provide them within this CRC, optimize them further and use them to study the evolution of gene expression in acute myeloid leukemia (AML) cells from patients and Patient-derived xenografts (PDX) cells grown in mice.
This will allow a comprehensive characterization of gene expression evolution in treated and untreated AML cells leading to a better understanding of phenotypic heterogeneity in cancer cells and how it evolves. This will eventually be informative for designing better treatment strategies.
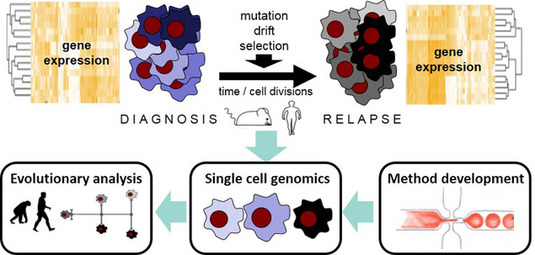
Figure legend: Characterizing gene expression evolution in AML using single cell genomics.

Prof. Dr. Wolfgang EnardFaculty of Biology, Ludwig-maximilians-Universität München +49 (0)89 2180-74339 |